 I normally do not put much personal information on this blog, but given today is International Rare Disease Day and I have experience with a very rare disease, I thought I would make an exception. In fact, this disease is so rare, it often appears in lists of the top ten rarest diseases (although my suspicion is that those lists are crap… but it is still extremely rare).
I normally do not put much personal information on this blog, but given today is International Rare Disease Day and I have experience with a very rare disease, I thought I would make an exception. In fact, this disease is so rare, it often appears in lists of the top ten rarest diseases (although my suspicion is that those lists are crap… but it is still extremely rare).
My issues first started in July 2010 when I began getting ulcerations on my tongue and cheeks. I initially thought that these were caused by biting in my sleep and figured that it would go away eventually. But it did not, and when I ended up spitting out more blood than toothpaste when brushing my teeth I decided it was time to see a doctor. The doctor I saw also thought it was bruxism and sent me to a dentist. The dentist decided that my wisdom teeth were a probable cause given where the damage in my cheeks was and that they were a bit out of line. So I had them pulled out and was put on various antibiotics to resolve any potential infection that could be involved. When I went back to see the dentist a couple of weeks after the second set of wisdom teeth were removed, he took a look in my mouth and decided there was something else going on.
I then was referred to an oral and maxillofacial surgeon. She took one look at my mouth and then got her receptionist to call an oral pathologist and get me an urgent appointment. Urgent, not because she though it was going to kill me, but because it looked quite bad (basically a direct quote…). She also sent me to get blood tests done to rule out a wide variety of infectious diseases. I did find it kind of amusing that she actually asked me if it was fine to test for some STDs rather than just saying this is what you are being tested for… Anyway, unsurprisingly I was all clear on that front.
Then I was onto seeing the oral pathologist. He spent a lot of time holding out my lip and staring at it. By this time I was starting to get blisters on the inside of my bottom lip and on the roof of my mouth. On the first visit he took two hole-punch biopsies from my mouth – and they are what they sound like… Your mouth gets numbed and a “small” cylinder of tissue is removed. This is also when I started on prednisolone (but more on that later…). Those biopsies came back suggesting I had an autoimmune disorder called lichen planus. The level of prednisolone I was taking was increased but this made little difference. I am fairly certain that the oral pathologist was not convinced of the diagnosis because he did much more lip staring and decided more biopsies were needed. Those came back as general ulcerated tissue (i.e. completely non-diagnostic) and so he sent me back to the oral surgeon so she could take a larger sample. Again, this was non-diagnostic as the tissue was just too damaged to get a clear view of which layer of the dermis the blistering was occurring in. By this stage, I had started getting blistering on my skin so I was sent to a dermatologist.
The dermatologist took a look at me and decided it was most likely a form of oral lichen planus that I had. But to make sure, more biopsies were taken from my lip… As the prednisolone was not helping a lot, it was time to get me onto some immune suppressing medication. That sort of medication is quite expensive so I was brought into the public hospital system where the doctor could apply to get the medication at a greatly subsidized rate. I was started on cyclosporin in February 2011. A biopsy from my finger (local anesthetic to the hand is really painful!) and some antibody tests appeared to confirm the diagnosis of lichen planus. Some antibody levels that would indicate another auto-immune disease affecting the skin were slightly raised, but not enough to change the diagnosis as these levels would not be unexpected given all the damage I had to the skin.
Around this time I started noticing I was getting shortness of breath while exercising. As I was told, you are suppose to get a bit puffed while doing exercise… but it was different. The best I could explain it was that I was feeling wheezy. So I was referred to the thoracic department where I had breathing tests and an ECG done. From those results, I was scheduled to have an echocardiogram and a CT scan of my chest.
While this was happening, there was not much progress in the healing of my mouth, so I was put on acitretin – a drug normally used to treat psoriasis, but also shows some evidence of helping lichen planus. And things did start to improve while on both medications.
I went for the echocardiogram and, from what I now know of how a normal one goes, things were not right. The ultrasound person had their supervisor come in and I was scanned while drinking water. A few days later I received a phone call saying the echo has shown a large mass behind my heart (“fist sized”) and I really should not miss the CT scan in the next couple of days. Also they were arranging for me to be admitted to hospital right afterwards so they could arrange further treatment. At this time it was decided I would probably need an immune system to deal with whatever that mass was, and given the acitretin appeared to be working, I was taken off the cyclosporin.
The CT scan did not give a clear diagnosis about what the mass was, although it did show it was not attached to my esophagus, which was good as I knew from various seminars I attended for work that cancer there is very high on the “not a good thing” scale. While I was in hospital I had a bronchoscopy performed with biopsies taken from my lung, which did not provide anything diagnostic to why I was having difficulty breathing. There was some Pneumocystis bacteria seen at levels that were not really a concern. I was brought back in the next week for an endoscopy where they were going to punch a small hole through my esophagus to get a sample from the mass. That was aborted mid-procedure as instead of having just a small hole punched through the esophagus, a tear occurred instead. The doctor doing the endoscopy had not seen anything like that before.
It was decided to just go in and look at the mass directly and, if it seemed a good idea at the time, to then remove it. So I was scheduled for surgery a couple of weeks later, which was late May 2011. During that two week period, I got an extensive flare-up of the blistering of my mouth and what looked like conjunctivitis in my eyes. When I say extensive, it was really extensive… the entire lining of my cheeks and roof of my mouth was gone and my tongue has extensive ulceration. The best way I have of describing it is to think of the blisters you get on the roof of your mouth when you eat a hot slice of pizza. Now extend them to everywhere. I completely lost all skin on my lips and started getting blistering over my face and ears. The blistering on my hands became a lot more extensive and my fingernail beds became swollen and the base of the nails became thin to the point of not being there. I also had an itchy rash covering most of my body. My endocrine/bone doctor actually wrote on my chart that I looked “horrid” and that the issues I was having due to being on prednisolone for the last eight months (low testosterone levels, bone density dropping…) were the least of my problems. I was also starting to lose a lot of weight given I had difficulty eating anything substantial, but luckily I had backup weight so that never became too much of an issue. I spent a few nights in hospital being put on various drips and having tests done to make sure I got to the surgery in a reasonable condition.
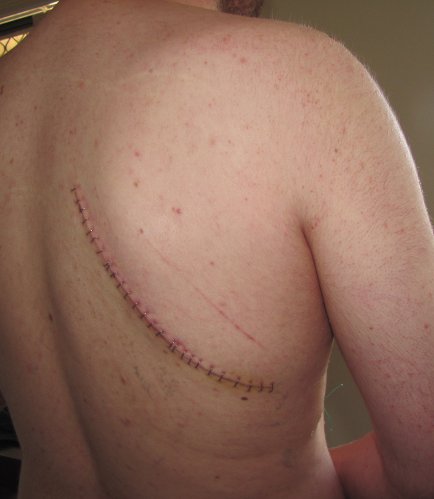
When I went to hospital for the surgery to remove the mass, the surgeon took a look at me and became very concerned to the point where I think he was considering delaying. I think the fact that I was not going to get any better without going on immune suppression again and that it would be better if we knew what the mass was before that happened is what convinced him to go ahead (and probably my wife crying had an influence…). The surgery was successful, with the entire mass removed and leaving me with an awesome looking scar (there are 26 staples there). The doctors could not tell what the mass was when they removed it but a week or so later it came back as being Castleman’s disease. That diagnosis in itself was quite good as removal is usually curative. And while that is very rare, I can still do “better”!
{kind=link}
It was with this diagnosis that things started to get placed together. When I saw the dermatologist next he took one look at me and admitted me to hospital. I definitely did not have lichen planus… Taking the Castleman’s into account, it was very likely I had paraneoplastic pemphigus. Another couple of biopsies from my hand and more antibody tests and this was confirmed. That antibody that previously had levels slightly elevated above normal was now off the charts so there was no doubt in the diagnosis. This was also confirmed by the eye symptoms I was having which on close inspection showed the surface of the eye eroding, which is common with paraneoplastic pemphigus.
I was going to need some quite extreme immune suppression. The short term treatment is extremely high doses of prednisolone and all its side effects… But before doctors hammer your immune system to the extent needed, you are checked for every infection they can think of. The infectious diseases team came and saw me and asked questions about my lifestyle that I never think I will be asked again! Anyway, I was once again cleared of all infectious disease tested for. The only concern was the bacteria observed during the bronchoscopy as it can cause a type of pneumonia in people with reduced immune systems. So once a treatment for that was sorted out (because I am potentially allergic to the usual medication), I was back on immune suppressants; this time mycophenolate. After a bit more than two weeks in hospital, it was decided that I no longer needed daily testing and I could manage my treatment at home.
I was out of hospital for a couple of weeks and then had my lungs tested again. Since my previously measurement, there was quite a substantial drop in lung function (for example, the FEV1 measure was now around a third of the expected value). Combined with the diagnosis of Castleman’s disease and paraneoplastic pemphigus, this lead to the diagnosis of bronchiolitis obliterans, which takes the entire situation from being awful to life threatening. In bronchiolitis obliterans, the small airways in your lungs become scarred which prevents the flow of air through them (from what I understand, it is mainly the outwards flow that is affected). I describe it as being like having a permanent severe asthma attack, although that might be entirely incorrect medically.
So it was back to hospital for another two weeks while the best course of treatment was decided. Bronchiolitis obliterans is irreversible, so the best that could be hoped for was stopping the decline in lung function. The immune suppression I was already on is a good start at controlling this, so it was doubled. That also allowed the dose of prednisolone I was on to be dropped quite a bit (I was on 75mg a day, which is a very high dose, for over a month and I had all the usual side effects – moon face, rapid weight gain particularly around the stomach, severe acne, mood swings…). It was also decided that I should do a four week course of Rituximab, which is a type of chemotherapy, but one that specifically destroys B-cells so you do not get side-effects people tend to think of with chemotherapy. I am also given a dose of IVIG monthly.
While this was going on, I was referred to a lung transplant doctor. With how my lungs are, there is not much room for further damage to be done before I would have significant difficulty doing every day tasks. There is only one case in the literature of someone having a lung transplant due to the follow-on effects of Castleman’s disease, so there is not much information to go on. The summary of my meetings with the lung transplant people was that I would not be a good candidate if my condition continued getting worse because there was nothing to say that my immune system would not just destroy the “new” set of lungs too. But if my condition stayed stable, I would have enough lung function to not warrant the risk of having a lung transplant now, particularly because living ten years with a lung transplant is a very good success. So ideally, the progress of my lung function decline due to the autoimmune condition would be halted now and I would only require a transplant later in life when the effects of the natural decline in lung function hits me earlier than most people because I will be starting from a worse base.
As my lungs no longer work very well and the sitting around in hospital and at home recovering did nothing to help my fitness, I was sent to a pulmonary rehabilitation program. This is a combination of cardio training and muscle building (as the fitter you are, the better you can handle low oxygen levels) and education (e.g. in how to manage shortness of breath, nutritional advise, some group counseling). Regaining fitness is particularly difficult when you have difficulty breathing on any moderate exercise. An interesting thing that I found out (although is really quite obvious when you think about it) is that the large muscle groups in your legs require a lot of oxygen, and they are used in everyday activities, so strengthening them can help you coping with shortness of breath. I also found out that a side-effect of being on high doses of prednisolone is muscle atrophy (wasting) so that would not have helped me in that regard.
And that about sums up where I am up to now. My lung function tests over the last six months have been quite stable so it looks like my current medication regiment is working. I have also noticed some improvement in my ability to do things like walk up a set of stairs without becoming short of breath, but I still have difficultly walking up hills or even walking fast on the flat. Hopefully keeping up with the exercise routine I am doing will see some more improvement, but there are really no guarantees there. My skin issues have mostly cleared up, leaving only a couple of small ulcerations on my tongue. The surface of my eyes also healed, but I am left with extremely dry eyes and have to continuously put in eye-drops. The only new issue I have had lately is an increased resting pulse rate (compensating for a lack of oxygen getting to my bloodstream). But multiple scans of my heart show there is no heart disease or any other issues so that is not too bad.
What can you do to help? There is not a lot that can be done for me personally, but I recommend the following:
1) Support medical research for rare diseases. Rare diseases are hard to diagnose, hard to treat and, as someone who does medical research, I know they are near impossible to get funding to do research into. If you see someone collecting for research into a rare disease and can spare a bit of money, give a donation.
2) Donate blood if you can. The monthly treatments of IVIG I receive comes from the plasma of a large number of donors. And like all blood products, there is a greater demand that what is donated, so help out if you can. Needles are not all that scary…
3) Sign up to donate your organs. The number of people needing organ donations far exceeds the number that donate. Your organs are not that much use to you once you are dead and with more people donating the organ matches will be better and the success rates higher. Also, tell your family that you want to donate your organs as with the current laws in most countries they will get the final say. I have always supported the idea of changing the law to make donation an “opt-out” process rather than the current “opt-in” and even removing the families right to override your decision.
And probably being more optimistic than anything…
4) Support stem cell research. I do not think the approach of “inject stem cells and hope for improvement” will work in my case but the growing of new organs from an individual’s own stem cells is a promising area of research and it would completely remove transplant rejections. Making a set of lungs would be very difficult, but recently a person had their trachea replaced with one built artificially from stem cells, so progress is being made.
5) Support artificial lung research. If I can not get a genuine pair of second-hand lungs in the future, then I will settle for being part machine. That would be cool!
Holy cow, that’s like a triple episode of House… I will try not to complain about minor health issues from now on!
An absolutely incredible set of events. The terminology alone is daunting. I wish you the best of luck with your continued recovery. Although I have already signed up to be an organ donor, I have not donated blood recently. This is certainly a great motivation. Thank you for sharing your story.
Hi Allan, it is important to raise the general awareness of rare diseases. I only learned about them because my local university opened a “centre for rare diseases”. Your post is really remarkable!